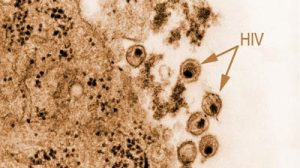
Neu gebildete Viren werden abgefangen
Neben dem Immunsystem kann der Körper weitere Schutzmechanismen aktivieren, um Infektionen mit Viren zu bekämpfen oder zu stoppen: Die befallenen Zellen selbst verfügen über eine Reihe von Eiweißen, die verschiedene Schritte der Virusvermehrung behindern.
In Gegenwart des Schutz-Proteins CD317 werden neu gebildete Viren beim Verlassen der Zelle fest an die Zelloberfläche gebunden und dies behindert sie am Befall weiterer Körperzellen. HIV überwindet diese Blockade mit seinem Protein Vpu: Es setzt diesen Schutzmechanismus, der interessanterweise gegen eine Vielzahl von Virenarten wirkt, gezielt außer Kraft.
Vpu stört Schutz-Protein
Das Virologenteam um Keppler untersuchte in einer Studie, auf welche Weise Vpu das Schutz-Protein CD317 in seiner Wirkung stört. Sie stellten fest, dass in menschlichen Zellen, in denen nach HIV-Befall der Störfaktor Vpu gebildet wird, der Vorrat an CD317 auf rund ein Viertel der ursprünglichen Menge zusammenschrumpft. „In Anwesenheit von Vpu wird CD317 von einem zelleigenen System zügig abgebaut. Vermutlich bindet Vpu an CD317 und markiert es für eine schnelle Beseitigung“, erklärt Keppler.
Je weniger CD317 in der Zelle vorhanden ist, desto mehr Viren können der Abfangvorrichtung entkommen. „Diese Interaktion zwischen Vpu und CD317 zu stören, wäre daher ein vielversprechender therapeutischer Ansatz, um die zelleigenen Schutzmechanismen zu stärken“, so Keppler.
Auch Ratten und Mäuse verfügen über dieses Schutz-Protein. Es übernimmt dieselbe Funktion und ist in der Lage, das HI-Virus des Menschen zu blockieren. Es gibt allerdings einen wichtigen Unterschied: Die Heidelberger Virologen entdeckten, dass Vpu dem CD317 in Rattenzellen nichts anhaben kann. „Das HI-Virus ist auf den Menschen spezialisiert, der Störmechanismus durch Vpu kann daher gegen den tierischen Infektionsschutz nichts ausrichten“, sagt Dr. Christine Goffinet, Erstautorin der Studie.
Ratten-Tiermodell soll nun verbessert werden
Dieses Detail ist wichtig, wenn die HIV-Infektion im Tiermodell an Ratten nachgestellt und untersucht werden soll: Die Infektion nimmt in der Ratte nicht denselben Verlauf wie im Menschen, da wegen der intakten zellulären Schutzvorrichtungen deutlich weniger Viren freigesetzt werden.
Dank der neuen Forschungsergebnisse hoffen die Heidelberger Wissenschaftler nun, das Tiermodell zu verbessern. Ziel ist es, in den Ratten CD317 gentechnisch auszuschalten und somit einen Grad der HIV-Infektion zu erzielen, der dem des Menschen ähnlicher ist.
Bereits 2007 war es den Heidelberger Wissenschaftlern gelungen, Ratten durch gezielte Veränderung ihres Erbmaterials erstmals für eine Infektion mit HIV empfänglich zu machen. An diesen „transgenen“ Ratten testeten sie erfolgreich Medikamente gegen die HIV-Infektion beim Menschen. Mit diesem transgenen Kleintiermodell ist es möglich, die Wirksamkeit von Medikamenten gegen den AIDS-Erreger HIV schnell und in größerem Umfang vor ihrem Einsatz beim Menschen zu prüfen und so die Weiterentwicklung neuer Virostatika zu beschleunigen.
(idw – Universitätsklinikum Heidelberg, 07.04.2009 – DLO)
7. April 2009