
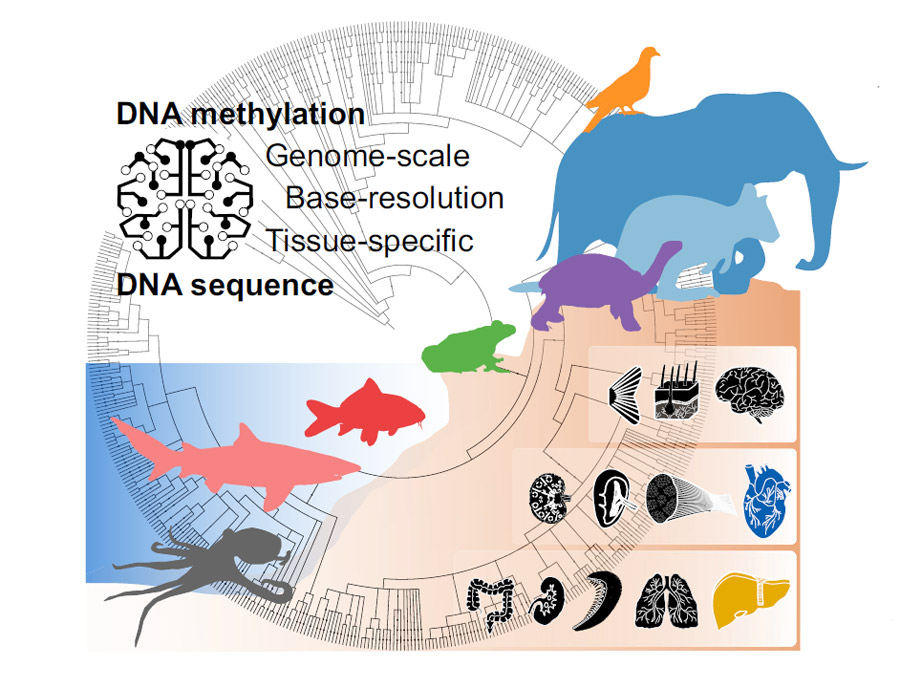
Vom Oktopus zum Elefanten: Forschende haben erstmals das Muster der DNA-Anhänge bei 580 Tierarten vergleichend kartiert – und so einen Stammbaum des Epigenoms erstellt. Die Kartierung dieses „zweiten Codes“ des Lebens enthüllt, dass sich die epigenetischen Muster selbst zwischen sehr unterschiedlichen Arten gleichen und warum. Sie zeigen aber auch, dass es in der Evolution der Wirbeltiere zwei auffallende „Sprünge“ im Epigenom gegeben hat. Die Ergebnisse bilden nun eine Referenz für weitere Studien – unter anderem zu krankmachenden epigenetischen Veränderungen.
Sie bilden den „zweiten Code“ des Lebens: Epigenetische Anlagerungen an unserer DNA kontrollieren, welche Gene abgelesen werden und welche stummgeschaltet sind. Sie bilden damit gewissermaßen die „Software“, die die Aktivität unserer genetischen „Hardware“ reguliert. Das Epigenom ist es auch, das unsere Zellen trotz identischer DNA zu völlig verschiedenen Zelltypen und Geweben werden lässt. Prägend dafür ist vor allem die Methylierung – Anhängsel blockierender Methylgruppen (-CH3) an den DNA-Basen.
„Die DNA-Methylierung ist das Gedächtnis der Zellen und sorgt dafür, dass eine Leberzelle immer eine Leberzelle und eine Herzzelle eine Herzzelle bleibt – obwohl alle Zellen unseres Körpers mit den gleichen Genen ausgestattet sind“, erklärt Seniorautor Christoph Bock von der Medizinischen Universität Wien. Außerdem spielt die DNA-Methylierung eine entscheidende Rolle für unsere Gesundheit – sie beeinflusst das Krebsrisiko und das biologische Alter, macht uns zu Sportmuffeln und verrät sogar, ob wir ein Zwilling sind.
580 Tierarten im Vergleich
Doch wie hat sich das Epigenom im Laufe der Evolution entwickelt? Teilen andere Wirbeltiere grundlegende Muster unserer DNA-Methylierung? Das hat nun ein Team um Bock und Erstautorin Johanna Klughammer vom CeMM Forschungszentrum für Molekulare Medizin der Österreichischen Akademie der Wissenschaften erstmals umfassend untersucht. Dafür analysierten die Forschenden Proben verschiedener Organe und Gewebe von 580 Tierarten – vom Oktopus bis zum Elefanten.
Zusätzlich untersuchten die Forschenden für 207 dieser Tierarten, ob und wie sich das Muster der DNA-Methylierung zwischen verschiedenen Geweben und Organen wie Lunge und Leber unterscheidet. Dahinter stand die Frage, ob beispielsweise das Lungengewebe eines Frosches epigenetisch der menschlichen Lunge ähnlicher ist als einem anderen Froschgewebe.
DNA-Code und Epigenom sind eng verknüpft
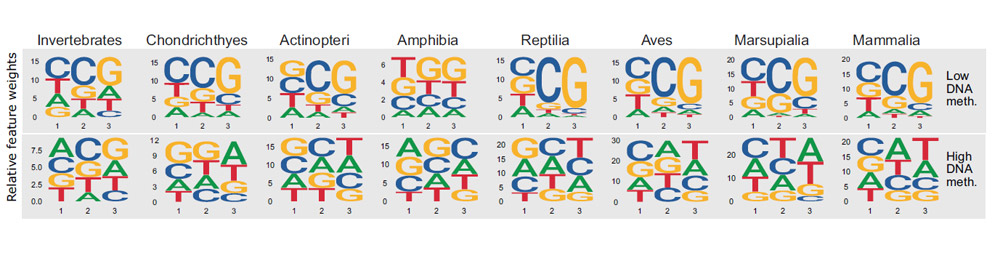
Die Epigenom-Kartierung enthüllt: In rund 500 Millionen Jahren der Evolution ist das epigenetische Grundmuster überraschend gleichgeblieben. Wo sich die Methylgruppen anlagern, ist demzufolge kein Zufall, sondern wird auch durch den DNA-Code mitbestimmt. Bestimmte Basenabfolgen sind demnach „anfälliger“ für eine Anlagerung als andere. „Dieses Muster war durch alle taxonomischen Gruppen hindurch erhalten“, berichten die Forschenden. „Dies stützt die Existenz eines genomischen Codes – einer prädiktiven Beziehung zwischen der DNA-Sequenz und der DNA-Methylierung.“
Die DNA-Methylierung in Seesternen und Fischen folgt demnach einem ganz ähnlichen Prinzip wie beim Orang-Utan oder bei uns Menschen. „Wir konnten zum Beispiel die Verteilung der DNA-Methylierung im Erbgut des Elefanten mit einem Modell vorhersagen, das wir für den Oktopus erstellt hatten“, sagt Klughammers Kollegin Daria Romanovskaia. „Diese epigenetischen Muster gab es folglich sehr wahrscheinlich schon beim letzten gemeinsamen Vorfahren dieser Tiere.“
Organe zeigen artübergreifend ähnliche Muster
Die Forschenden vermuten, dass dieser grundliegende Code eine entscheidende Rolle unter anderem für die Differenzierung der Gewebe im Embryo spielt. Dazu passt, dass viele organ- und gewebespezifische Signaturen artübergreifend sehr ähnlich sind. Einige Anlagerungsmuster sind demnach beim gleichen Organ verschiedener Tierarten ähnlicher als bei verschiedenen Geweben desselben Tieres. Dies galt vor allem bei Knochenfischen, Vögeln und Säugetieren.
Das evolutionär konservierte Muster von sequenz- und organspezifischen Methylierungen könnte nach Ansicht des Teams eine wichtige Rolle unter anderem für die Differenzierung und Entwicklung des Embryos spielen. „Ein Art Default-Zustand des Epigenoms, der in der DNA-Sequenz selbst kodiert ist, könnte die Ausgangslinie repräsentieren, die dann im Laufe des Lebens durch andere Effekte modifiziert wird“, erklären Klughammer und ihre Kollegen.
Zwei große Sprünge im Stammbaum
Dennoch gibt es klare Unterschiede und Veränderungen im Laufe der Evolution: Das Team identifizierte unter anderem zwei deutliche Sprünge im epigenetischen Stammbaum. „Der genetische Code der Epigenetik ist in Wirbeltieren klarer und verbindlicher als in Wirbellosen, auch wenn die zugrundeliegenden Muster ähnlich sind“, berichtet Bock. „Und mit dem Aufkommen der Reptilien, Vögel und Säugetiere wird die genetische Komponente der DNA-Methylierung noch ausgeprägter.“
Ein Zusammenhang besteht dabei auch mit der Größe und Langlebigkeit der Tierarten: Weil solche Organismen ein höheres Risiko unter anderem für die Entartung von Zellen haben, scheint ihr Genom besonders gut durch DNA-Anlagerungen geschützt und kontrolliert zu sein. Je höher das theoretische Krebsrisiko einer Tierart war, desto höher war auch die DNA-Methylierung, wie das Team feststellte. Obwohl ein Elefant von Natur aus ein höheres Krebsrisiko haben müsste, erkrankt er dadurch nicht häufiger an Krebs als die kleine, kurzlebige Maus oder eine Forelle.
Referenz für weitere Forschung
Insgesamt liefert die Studie den bisher umfassendsten Stammbaum des tierischen Epigenoms und liefert damit wichtige Einblicke in die biologischen Muster und Grundprinzipien, die diesen „zweiten Code“ des Lebens prägen. Die daraus gewonnenen Erkenntnisse können nun einen Ausgangspunkt für tiefergehende Studien bilden, die beispielsweise gezielter bestimmten Unterschieden zwischen Mensch und Tier nachgehen, aber auch den Faktoren, die das Epigenom verändern.
Im Rahmen ihrer Studie haben die Forschenden zudem neue Analysemethoden entwickelt, durch die die DNA-Methylierung auch unabhängig von einem Referenz-Genom für die jeweilige Tierart untersucht werden kann. „Dies ermöglicht es, das Zusammenspiel von Genetik und Epigenetik all jener Tierarten zu studieren, die bisher für epigenetische Analysen kaum zugänglich waren“, erklärt Klughammer. „Und hoffentlich können wir so auch die Rolle der Epigenetik beim Menschen, bei der Entstehung von Krebs und beim gesunden Altern besser verstehen.“ (Nature Communications, 2023; doi: 10.1038/s41467-022-34828-y)
Quelle: Nature Communications, CeMM Forschungszentrum für Molekulare Medizin der Österreichischen Akademie der Wissenschaften











