Am Anfang des Sehvorgangs steht die Wechselwirkung des Lichts mit dem Protein Rhodopsin. Dieses enthält den eigentlichen Lichtsensor, der angeregt wird, seine Form zu verändern und so den Rest des Vorgangs anzustoßen. Forscher haben nun die Struktur des Rhodopsinmoleküls in dem kurzlebigen angeregten Zustand bestimmt und so ein präzises Bild davon geliefert, was in der Netzhaut am Anfang des Sehvorgangs geschieht.
Dieses Ergebnis dürfte auch zu einem besseren Verständnis der erblichen Augenkrankheit Retinitis Pigmentosa beitragen und möglicherweise Wege für deren Behandlung oder Verlangsamung aufzeigen, schreiben die Wissenschaftler in der Online-Ausgabe von „Nature“. Gleichzeitig liefert die Studie die Basis für das Verständnis vieler weiterer Vorgänge im Organismus, die auf einem ähnlichen Mechanismus beruhen – etwa die Wahrnehmung von Gerüchen oder die Steuerung von Abläufen über Hormone.
Hochkomplexer Vorgang
Sehen ist ein hochkomplexer Vorgang – eine Vielzahl von chemischen Reaktionen muss ablaufen bevor das Gesehene unser Bewusstsein erreicht. Ganz am Anfang dieses Vorgangs trifft das Licht auf die Sehsinneszellen in der Netzhaut des Auges – die Zapfen oder Stäbchen. In den Zellmembranen der Stäbchen, die für das Sehen bei schlechten Lichtverhältnissen zuständig sind, sitzen Rhodopsin-Moleküle – die eigentlichen Lichtsensoren. Sie bestehen aus jeweils insgesamt sieben stabförmigen Molekülteilen, die von außen ins Innere der Zelle hineinreichen.
Fällt Licht von außen auf das Rhodopsin, verändert sich die Anordnung der stabförmigen Teile so, dass im Inneren der Zelle ein so genanntes G-Proteinmolekül Platz dazwischen findet. Das Andocken des G-Proteins stößt eine Kaskade von Vorgängen an, an deren Ende ein Nervenimpuls ausgelöst wird. Das eigentlich lichtempfindliche Pigment ist das Retinal – eine Form von Vitamin A – das als kleines geknicktes Molekül zwischen den sieben Teilen des Rhodopsins steckt. Sobald Licht darauf fällt, streckt es sich und drückt Teile des Rhodopsins auseinander, so dass Platz für das G-Protein entsteht.

Wie der Sehvorgang im Auge beginnt
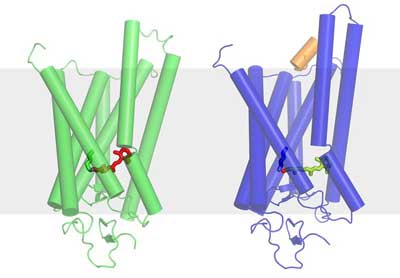
Nun ist es Forschern des Paul Scherrer Instituts (PSI) zusammen mit Kollegen aus Großbritannien und den USA gelungen, die Struktur des Rhodopsins im aktivierten Zustand zu bestimmen – also in der durch das Licht veränderten Form mit dem gestreckten Retinal. Dieser Zustand ist eigentlich sehr kurzlebig, da das Rhodopsin ja möglichst schnell in den Zustand zurückkehren muss, in dem es für Licht empfänglich ist.
Die PSI-Forscher haben aber einen Weg gefunden, das Molekül geringfügig so zu verändern, dass es die aktivierte Form länger beibehält und konnten damit seine Struktur bestimmen. Die Struktur der inaktiven Form des Rhodopsins, wie sie ohne Licht auftritt, war schon vorher bekannt. Mit der Kenntnis beider Strukturen kann man jetzt genau nachvollziehen wie der Sehvorgang im Auge auf molekularer Ebene beginnt.
Moleküle in einer Kristallstruktur angeordnet
Für die Untersuchungen erzeugten die Wissenschaftler die entsprechenden Moleküle in großer Menge und ordneten sie in einer Kristallstruktur regelmäßig an. Dabei ist Rhodopsin eines der sehr wenigen Membranproteine dieser Klasse, die sich kristallisieren lassen. Die Kristalle wurden mit Synchrotronlicht durchleuchtet und aus der Ablenkung des Lichts auf dem Weg durch den Kristall können die Forscher auf die Struktur der untersuchten Moleküle schließen. Die Messungen wurden an der Synchrotron Lichtquelle Schweiz SLS des PSI und an zwei weiteren ähnlichen Anlagen durchgeführt.
Universelle Mechanismen des Lebens verstehen
„Die Untersuchung des Rhodopsins hilft uns eine große Klasse von ähnlichen Molekülen zu verstehen – es gibt mehr als 800 davon im Menschen“, erklärt Jörg Standfuss, Leiter des Forschungsprojekts. „Die meisten reagieren nicht auf Licht, sondern auf andere Reize und erfüllen so die unterschiedlichsten Aufgaben: Im Geruchssinn reagieren sie auf Substanzen aus der Atemluft. Oder sie dienen als Rezeptoren für Hormone innerhalb des Körpers – wie etwa die Beta-Rezeptoren, die am Herzen für Steuerung des Blutdrucks mitverantwortlich sind“. Diese dienen als Andockstelle für die als Betablocker bekannten Mittel gegen Bluthochdruck.
Insgesamt sind diese Moleküle von großem Interesse für die pharmazeutische Forschung, weil man über sie Vorgänge im Körper sehr gezielt steuern oder blockieren kann. So wechselwirken etwa Medikamente, die bei Herzrhythmusstörungen, Migräne oder Allergien eingesetzt werden, mit diesen Rezeptoren. Der genaue Aufbau der Beta-Rezeptoren war Thema einer weiteren Arbeit, die PSI-Forscher mit Kollegen in Cambridge vor kurzem in Nature veröffentlicht haben.
Optimierte Therapien für Augenkrankheit
„Unsere Erfahrung mit der Strukturuntersuchung an veränderten Rhodopsin- Molekülen wenden wir derzeit auch zur Erforschung einer verbreiteten Augenkrankheit an – der Retinitis Pigmentosa“, erklärt Standfuss. Bei dieser ererbten Krankheit ist oftmals das Rhodopsin in den Zapfen des Auges verändert So wird es nicht wie im gesunden Auge regelmässig vollständig erneuert – es verbleiben stets Teile der „alten“ Moleküle, die allmählich die Sehzellen vergiften. Das führt anfangs zu Nachtblindheit und über längere Zeit zu einem deutlich eingeschränkten Gesichtsfeld.
Standfuss dazu: „In Zukunft werden wir genau bestimmen können, in welcher Weise das Rhodopsin bei der Erkrankung verändert ist, und dann auch untersuchen, wie kleine Moleküle, die als Medikamente die Erkrankung aufhalten, in das Rhodopsin eingebaut werden.“ Mit diesem Wissen könnte man dann am Computer die Struktur der Medikamente gezielt optimieren. (Nature, 2011; doi:10.1038/nature09795)
(Paul Scherrer Institut (PSI), 10.03.2011 – DLO)