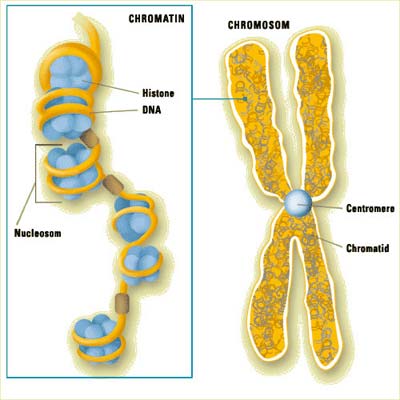
Das Genom von Lebewesen mit Zellkern weist eine charakteristische Struktur auf: Die fadenförmige DNA wickelt sich sehr kompakt zu so genannten Nukleosomen auf, die durch frei zugängliche DNA-Abschnitte verbunden sind. Diese Struktur bestimmt entscheidend mit, welche Gene an- oder ausgeschaltet werden. Münchener Biophysiker haben nun ein Modell entwickelt, mit dessen Hilfe sich die Verteilung der Nukleosomen um die strategisch wichtigen Startstellen der Transkription beschreiben lässt – die frei von Nukleosomen sein müssen.
{1r}
Die Transkription bezeichnet die Abschrift der Gene, also den ersten Schritt in der Übertragung genetischer Information in Proteine. Dabei entdeckten die Wissenschaftler um Professor Ulrich Gerland und Wolfram Möbius von der Universität München (LMU), dass sich auf beiden Seiten der nukleosomfreien Startstellen der Transkription unterschiedliche „Stopp“-Signale befinden, die die Bildung von Nukleosomen verhindern.
„Unser Modell könnte eine wichtige Hilfestellung bei der Entschlüsselung des so genannten Chromatincodes sein, der dem Genom seine Struktur verleiht und bisher noch weitgehend unverstanden ist“, sagt Gerland in der Fachzeitschrift „PLoS Computational Biology“.
Wie Perlen auf einer Kette
Das Erbmaterial höherer Organismen liegt eng gepackt in den Kernen ihrer Zellen. Die fadenförmige DNA wickelt sich dabei auch zu so genannten Nukleosomen auf. Deren Inneres besteht aus jeweils acht Histon-Proteinen, die – ähnlich einer Spule – von DNA umwickelt sind. Die Nukleosomen sind durch unverpackte DNA-Bereiche verbunden und erinnern dadurch an aufgefädelte Perlen. Sie bringen die DNA aber nicht nur in eine kompakte Form für die Verpackung in den Zellkern. „Sie beeinflussen auch, welche Bereiche des Erbmoleküls abgelesen und damit in Proteine übersetzt werden können“, erklärt Gerland.
Die Zugänglichkeit der DNA ist ein wichtiger Aspekt der Genexpression und für die Forschung von großem Interesse. Ein Schwerpunkt ist dabei die Frage, wie die Nukleosomen um die Startregionen der Transkription verteilt sind. Denn an diesen Stellen beginnt das Ablesen der DNA, das ist der erste Schritt in der Übertragung genetischer Information in Proteine.
An diesen Start- oder Promoter-Regionen wurde häufig ein typisches Muster beobachtet, bei dem eine nukleosomfreie Zone von Bereichen mit einer charakteristischen Verteilung von Nukleosomen umgeben ist. Die biologische Funktion dieser Lücken ist offenbar, der aus vielen Untereinheiten bestehenden molekularen Transkriptionsmaschinerie Andockstellen freizuhalten.
Physikalisches Modell erklärt Nukleosomen-Verteilung
Zusammen mit Möbius untersuchte Gerland nun, ob ein einfaches physikalisches Modell die Verteilung der Nukleosomen rund um eine Promoter-Region erklären kann. Die Forscher nutzten dafür das so genannte Tonks-Modell, das die Wechselwirkung zwischen Gaspartikeln beschreibt, wenn sich diese nur in einer Dimension bewegen können.
„Kennt man die Position eines Teilchens, kann man mithilfe des Modells die Positionen der anderen Teilchen vorhersagen“, sagt Möbius, der Erstautor der Studie. „Zudem lassen sich charakteristische Oszillationen zwischen den Gaspartikeln beobachten.“ Die Analysen der beiden Forscher ergaben, dass das Tonks-Modell auch die Verteilung der Nukleosomen erstaunlich gut beschreiben kann.
„Wenn wir die Durchschnittswerte vieler verschiedener Promotor-Regionen einsetzen, gibt das Modell die nukleosomfreie Zone mit der typischen Schwankung der Nukleosom-Dichte auf beiden Seiten wieder“, berichtet Gerland. Das neue Modell stimmt am besten mit den Daten überein, wenn für beide Grenzregionen der nukleosomfreien Zone unterschiedliche Randbedingungen angenommen werden.
Nukleosom als „Straßenblockade“
„Auf der einen Seite – und zwar in Richtung der Transkription – muss es ein Nukleosom geben, das wie eine Art Straßenblockade die Lücke ohne Nukleosomen freihält“, meint Gerland. „Auf der anderen Seite, also gegen die Transkriptionsrichtung, muss dagegen ein breiter, gewissermaßen abstoßender Bereich vorliegen. Wie eine Art Verkehrsschild muss er signalisieren, dass sich hier keine Nukleosome bilden.“
Modell von Roger Kornberg bestätigt
Die Forscher konnten mit dem vorliegenden Ergebnis erstmals das Modell des amerikanischen Nobelpreisträgers Roger Kornberg quantitativ bestätigen, der 1974 die Nukleosomen entdeckte und später ein Modell für ihre statistische Verteilung im Genom entwickelte. Das neue Modell könnte wesentlich dazu beitragen, die Regeln zu verstehen, nach denen die Struktur der Chromosomen festgelegt wird.
„Unsere Berechnungen könnten eine Hilfestellung bei der Dekodierung des so genannten Chromatincodes sein, über den bisher noch wenig bekannt ist“, sagt Gerland. „In diesem Code ist festgelegt, wie das Genom seine dreidimensionale Struktur erhält.“
(idw – Universität München, 23.08.2010 – DLO)