Auch unter den „orphan diseases“ gibt es einige Krankheiten, die in ihrer Seltenheit hervorstechen und mit zum Teil höchst außergewöhnlichen Symptomen erstaunen. Manche dieser Krankheiten betreffen nur einige hundert Menschen weltweit und bei anderen gibt es sogar nur eine Handvoll Betroffene.
Fibrodysplasia ossificans progressiva
Diese seltene Krankheit geht mit einer fortschreitenden Verknöcherung von Strukturen des Binde- und Muskelgewebes einher – Betroffenen wächst sozusagen ein zweites Skelett über das Erste. Schon die kleinsten Gewebeverletzungen führen sofort zu einer Verschlechterung des Gesundheitszustands, da sich anstatt von neuen Gewebezellen neues Knochengewebe bildet. Bisher sind weltweit etwa 800 akute Fälle bekannt.
Eine einzelne Mutation im Gen eines bestimmten Rezeptors ist verantwortlich für das gesteigerte Knochenwachstum: Durch die Mutation kommt es zu einer Überaktivierung dieser Andockstelle, die sich auf vielen Körperzellen befindet und das Wachstum sowie die Entwicklung des Skelett koordiniert.
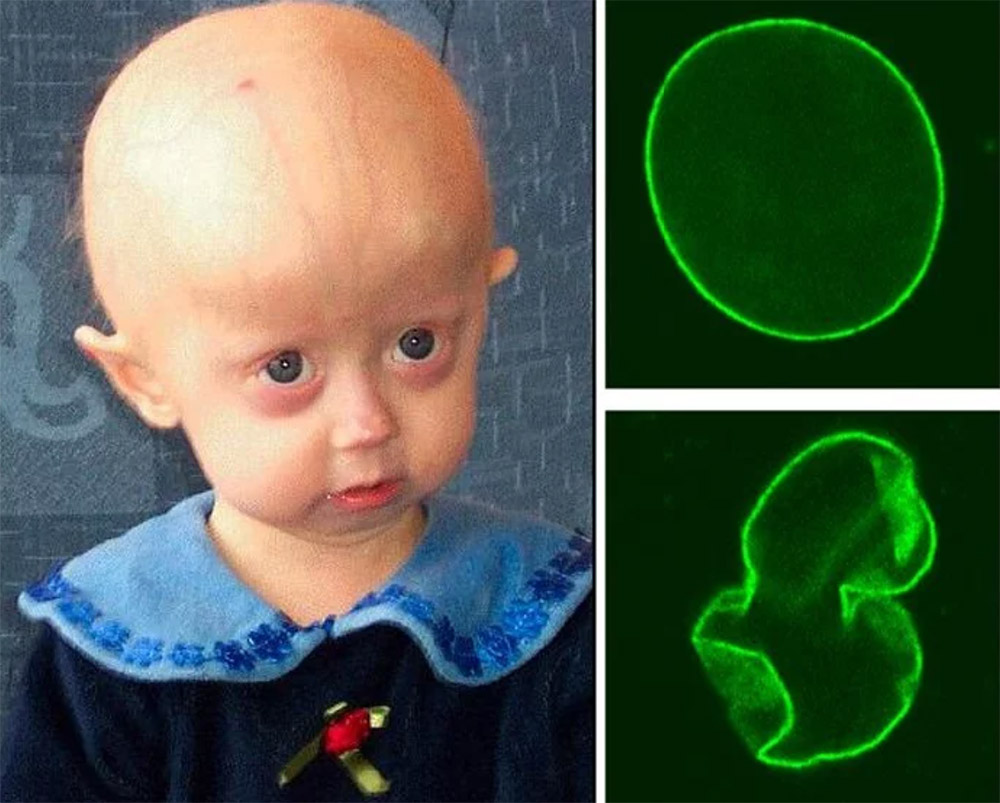
Hutchinson-Gilford-Progerie
Altern wie im Zeitraffer: Das ist nicht nur eine dystopische Vorstellung, sondern für etwa 200 Kinder auf der Welt die Realität. Sie altern etwa fünf bis zehnmal schneller als normale Menschen und sterben im Alter von circa 14 Jahren meistens an einem Herzinfarkt oder einem Schlaganfall. Eine Therapie um das beschleunigte Altern aufzuhalten gibt es bisher nicht.