Die Aufklärung der Proteinstruktur ist entscheidend, um die Funktion der Biomoleküle nachvollziehen zu können und die molekularen Mechanismen in unserem Körper weiter zu verstehen. Dies ist vor allem für die Medikamentenentwicklung wichtig, denn wie der englische Name „structure-based drug design“ schon verrät, basieren die meisten Arzneimittel auf der Struktur des Proteins, an dem ihre Wirkung ansetzen soll – sei es an einem körpereigenen Rezeptor oder wie bei Antibiotika, am Protein eines Erregers.
Ein Beispiel sind gängige Medikamente gegen HIV, die ein entscheidendes Enzym des Virus blockieren. Für die Entwicklung dieser Mittel muss zuvor die Struktur des Zielproteins aufgeklärt werden – in diesem Fall der HIV-Protease.

Strukturaufklärung ist eine knifflige Angelegenheit
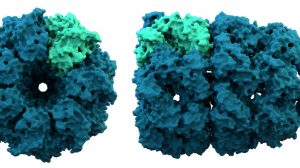
Die Aufklärung der dreidimensionalen Struktur eines Proteins erfolgte erstmals im Jahr 1958 durch John Kendrew, der die Struktur des Muskelproteins Myoglobin aufklärte. Diese Entschlüsselung der Faltung ist nur durch komplizierte und aufwendige Verfahren wie Röntgenkristallografie oder Kernspinresonanzspektroskopie (NMR-Spektroskopie) möglich. Die Strukturen der meisten Proteine in der Proteindatenbank PDB wurden mittels Röntgenbeugung an Proteinkristallen aufgeschlüsselt. Solche entstehen, wenn man eine Proteinlösung kontrolliert verdunsten lässt und somit die Proteinkonzentration in ihr langsam ansteigen lässt – die Proteine kristallisieren dann aus.
Die Proteinkristalle beugen Röntgenstrahlen auf eine charakteristische Art und Weise, die von Wissenschaftlern in ein Strukturmodell übersetzt werden kann. Doch die optimalen Bedingungen für eine Proteinkristallisation zu finden, folgt einem Trial-and-Error Ansatz und kann mitunter sehr lange dauern.