Es gibt möglicherweise neue Hoffnung für Patienten mit der tödlichen Erbkrankheit Chorea Huntington: Ein bisher zur Muskelentspannung eingesetztes Mittel hat in Versuchen an Mäusen das Fortschreiten der Krankheit deutlich verlangsamt. Das Medikament Dantrolen hemmte den Abbau von Nervenzellen im Gehirn. Außerdem verhinderte es bei den Mäusen die für Huntington typischen Lauf- und Bewegungsstörungen. Das berichten Wissenschaftler in der kommenden Ausgabe des Fachmagazins „Molecular Neurodegeneration“.
{1r}
„Unsere Ergebnisse deuten darauf hin, dass Dantrolen und ähnliche Hemmstoffe sich als Mittel eignen, um das Fortschreiten von Krankheiten wie Chorea Huntington zu bremsen“, sagen Ilya Bezprozvanny von der University of Texas und seine Kollegen. Dantrolen habe sowohl in Zellkulturen als auch in Versuchen mit Mäusen die für die Muskelsteuerung zuständigen Zellen im Gehirn vor der Zerstörung geschützt.
Chorea Huntington bisher nicht heilbar
„Bisher gibt es zwar einige Medikamente, die die Symptome der Chorea Huntington lindern können, aber ein Heilmittel, das die Krankheit stoppt oder rückgängig macht, existiert nicht“, schreiben die Forscher. Dantrolen wecke nun die Hoffnung, dass man zumindest ein Medikament entwickeln könne, das die fortschreitende Zerstörung von Gehirnzellen verlangsame.
Da Dantrolen bereits seit Jahrzehnten als Arzneimittel gegen krankhafte Muskelkrämpfe eingesetzt wird, sind seine Nebenwirkungen relativ gut bekannt. Es kann unter anderem Muskelschwäche und Atemstörungen auslösen. Wie stark diese Nebenwirkungen sind, wenn das Mittel über lange Zeit mit der gegen Huntington wirksamen Dosis eingenommen wird, müsse aber noch untersucht werden, betonen die Wissenschaftler. Zumindest bei den Mäusen habe das Mittel auch nach monatelanger Anwendung keine schwerwiegenden giftigen Nebenwirkungen gezeigt.
Chorea Huntington trifft fünf von 100.000 Menschen
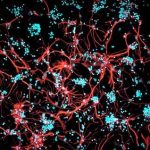
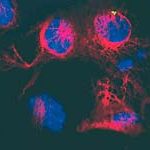
Die Krankheit Chorea Huntington gehört zu den häufigsten erblichen Nervenerkrankungen. In Deutschland sind nach Angaben von Forschern des Universitätsklinikums Charité in Berlin zwei bis fünf Menschen unter 100.000 betroffen. Bei diesen Patienten ist ein Gen auf dem vierten Chromosom verändert und produziert dadurch ein fehlgebildetes Eiweiß, das Huntingtin. Dieses Protein bildet schädliche Ablagerungen, die die muskelsteuernden Zellen im Gehirn zerstören.
Die ersten Symptome der Krankheit machen sich meist zwischen dem 30. und 40. Lebensjahr bemerkbar. Sie beginnen mit unwillkürlichen Muskelzuckungen und Bewegungsstörungen, denen die Krankheit auch ihren früheren Namen Veitstanz verdankt. Nach 15 bis 25 Jahren führt der fortschreitende Verlust der Muskelkontrolle letztlich zum Tod der Patienten.
Dantrolen im Mäusefutter verabreicht
Für ihre Studie nutzten die Forscher einen Mäusestamm mit krankhaft verändertem Huntingtin-Gen. Ein Teil dieser Mäuse erhielt von Jugend an Dantrolen mit dem Futter, eine Kontrollgruppe nicht. Auf gleiche Weise wurden zwei Gruppen von gesunden Wildtyp-Mäusen behandelt.
Bis zum Alter von 13,5 Monaten testeten die Wissenschaftler regelmäßig die Muskelkoordination der Mäuse, indem sie diese über dünne Balken balancieren ließen. Wie die Forscher berichten, schnitten die mit Dantrolen gefütterten Huntington-Mäuse hier genauso gut ab wie die gesunden Mäuse.
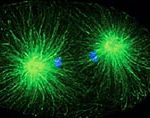
Nach Ende der Versuchsphase wurden die Mäuse getötet und ihre Gehirne untersucht. Bei den Dantrolen-behandelten Huntington-Mäusen habe man deutlich weniger abgelagertes Huntingtin-Eiweiß gefunden als bei den unbehandelten, sagen die Wissenschaftler. Das Mittel habe die Ansammlung des zellzerstörenden Proteins in den muskelsteuernden Zellen dramatisch reduziert. (Molecular Neurodegeneration, 2011)
(Molecular Neurodegeneration / dapd, 28.11.2011 – NPO)