Entzündungsreaktionen, die als Folge einer Gewebeverletzung entstehen können, gehen typischerweise mit einer erhöhten Empfindlichkeit gegenüber Schmerzen einher. Hierbei führen Reize, die bereits unter Normalbedingungen als schmerzhaft empfunden werden, zu einer massiv verstärkten Schmerzreaktion (Hyperalgesie), und sogar an sich neutrale Reize, wie beispielsweise leichte Berührungen, können heftige Schmerzen auslösen (Allodynie). Diese so genannte inflammatorische Sensitisierung gegenüber Schmerzreizen ist einerseits auf eine erleichterte Erregbarkeit der periphären Nervenzellen zurückzuführen. Forschungsarbeiten der letzten Jahre zeigen jedoch, dass andererseits auch Nervenzellen des Rückenmarks durch entzündliche Prozesse empfindlicher werden können.
Prostaglandine unter der Lupe
Doch welche molekularen Prozesse liegen diesem Phänomen nun zu Grunde? Hierbei scheinen Prostaglandine, eine Klasse von Botenstoffen, die während Entzündungsreaktionen ausgeschüttet werden, eine zentrale Rolle zu spielen. So beruht die Wirkungsweise von Schmerzmitteln, wie „Paracetamol“ oder „Aspirin“, auf der Hemmung der Prostaglandin-Synthese. Interessanterweise werden Prostaglandine, insbesondere der Subtyp E2 (PGE2), nicht nur von Zellen in der unmittelbaren Umgebung des Entzündungsherdes produziert, sondern auch im Rückenmark steigt der Prostaglandin-Spiegel massiv an. Das eingehende Schmerzsignal wird somit im Rückenmark über eine PGE2-abhängige Signalkaskade nochmals verstärkt. Diese Verstärkung erfolgt in den oberflächlichen Schichten des Rückenmarks, dem dorsalen Horn. Dieses ist die zentrale Schaltstelle zwischen schmerzempfindlichen Nervenfasern der Peripherie und nachgeschalteten Nervenzellen, die das Signal dann zum Gehirn weiterleiten.
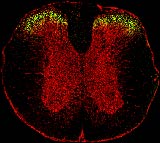
„Ziel unseres Forscherteams war es zunächst nicht,“ so Ulrike Müller, Projektleiterin am Max-Planck-Institut für Hirnforschung, „einen neuartigen Schmerzmodulator zu finden, sondern die Funktion eines Rezeptors für den Neurotransmitter Glyzin, der GlyRα3-Isoform, zu verstehen.“ Die Glyzin-Rezeptoren sitzen in der Zellmembran von Neuronen, wo sie porenartige Ionenkanäle ausbilden. Nach Bindung des Transmitters Glyzin öffnet sich der Kanal und Chlorid-Ionen strömen in die Zelle ein. Dieser Ionenfluss hemmt die Nervenreizleitung und dämpft das „Feuern“ der Neuronen. Um die Funktion von GlyRα3 zu verstehen, erzeugten die Forscher Antikörper gegen den Rezeptor, machten diese über Fluoreszenz-Farbstoffe sichtbar und inkubierten Gewebeschnitte des Rückenmarks von Mäusen mit diesen Antikörpern (vgl. Abb. 1).
Hemmung der Glyzin-abhängigen Nervenreizleitung
Diese Versuche zeigten für GlyRα3 eine spezifische Lokalisation im dorsalen Horn des Rückenmarks, was auf eine mögliche Funktion des Rezeptors in der Schmerzreizleitung hinweist. Aus elektrophysiologischen Untersuchungen der Arbeitsgruppe von Hanns-Ulrich Zeilhofer von der Universität Erlangen-Nürnberg wussten die Forscher, dass die Nervenimpulsleitung von Neuronen des dorsalen Horns durch Prostaglandine moduliert wird und dass hierbei Glyzin-Rezeptoren beteiligt sind. Mit Hilfe von genetisch modifizierten Mäusen, in denen das Gen für GlyRα3 inaktiviert wurde, haben die Forscher in Zusammenarbeit mit Zeilhofer gezeigt, dass spezifisch die α3-Isoform des Glyzinrezeptors von Prostaglandinen moduliert wird und diesen Rezeptoren damit eine Schlüsselfunktion in der inflammatorischen Schmerzsensitisierung zukommt.
So zeigten elektrophysiologische Messungen an Rückenmarksschnitten von GlyRα3-defizienten Tieren nicht mehr die typische Hemmung der Glyzin-abhängigen Nervenreizleitung. Um herauszufinden, welche Rolle GlyRα3 in der physiologischen Schmerzantwort tatsächlich hat, verglichen die Wissenschaftler die Schmerzempfindlichkeit von GlyRα3-defizienten mit der von normalen Mäusen. Beide Gruppen reagierten in gleicher Weise auf akute Schmerzreize. Induzierten die Wissenschaftler jedoch zunächst eine Entzündungsreaktion, zum Beispiel durch Injektion einer irritierenden Substanz in die Pfote, so konnten sie beobachten, wie die hierdurch in normalen Mäusen ausgelöste, langanhaltende Schmerzsensitisierung in den GlyRα3-defizienten Tieren ausblieb.
„Unsere Versuche zeigen also, dass der α3-Glyzinrezeptor essentiell ist für die nach einer Entzündung gesteigerte Schmerzreizleitung der Rückenmarksneuronen,“ so Müller weiter. Auf molekularer Ebene führt die Bindung des Prostaglandins PGE2 an Zelloberflächenrezeptoren zur Aktivierung einer Signalkaskade, die zur Übertragung von Phosphatgruppen auf den Rezeptor GlyRα3 führt. Hierdurch wird der Rezeptor inaktiviert und kann seine hemmende Wirkung auf die Schmerzreizleitung nicht mehr ausüben. Letztlich führt dies zu einer gesteigerten Aktivität der Rückenmarksneuronen, das Schmerzsignal wird somit verstärkt. „Gelänge es nun Substanzen zu entwickeln, die genau diesen Signalweg unterbinden, so könnte dies zu einer neuartigen Therapie für chronische Schmerzzustände führen“, sagt Müller.
(Max-Planck-Gesellschaft, 21.05.2004 – DLO)
21. Mai 2004