Genschere gegen Blutkrankheiten: In Großbritannien ist weltweit erstmals eine Therapie auf Basis der Genschere CRISPR/Cas9 zugelassen worden. Die Methode wird bei Sichelzellanämie und Beta-Thalassämie eingesetzt – beide Erbkrankheiten verursachen krankhafte Veränderungen des Blutfarbstoffs Hämoglobin. Die CRISPR-Therapie repariert jedoch nicht den ursächlichen Gendefekt, sondern reaktiviert die Produktion eines fötalen Hämoglobins. Dieses übernimmt dann die Funktion des defekten Blutfarbstoffs.
Die Sichelzellanämie und die Beta-Thalassämie sind beides Erbkrankheiten, die durch einen Defekt in der genetischen Bauanleitung für den Blutfarbstoff Hämoglobin verursacht werden. Als Folge werden fehlgebildete oder zu wenige rote Blutkörperchen gebildet. Dies führt zu schwerer Blutarmut, bei der Sichelzellanämie auch zu Schmerzen und Organschäden. Betroffene benötigen ein Leben lang regelmäßig Bluttransfusionen und Medikamente, längerfristige Abhilfe kann nur eine Knochenmarkspende und Stammzelltransplantation bringen.
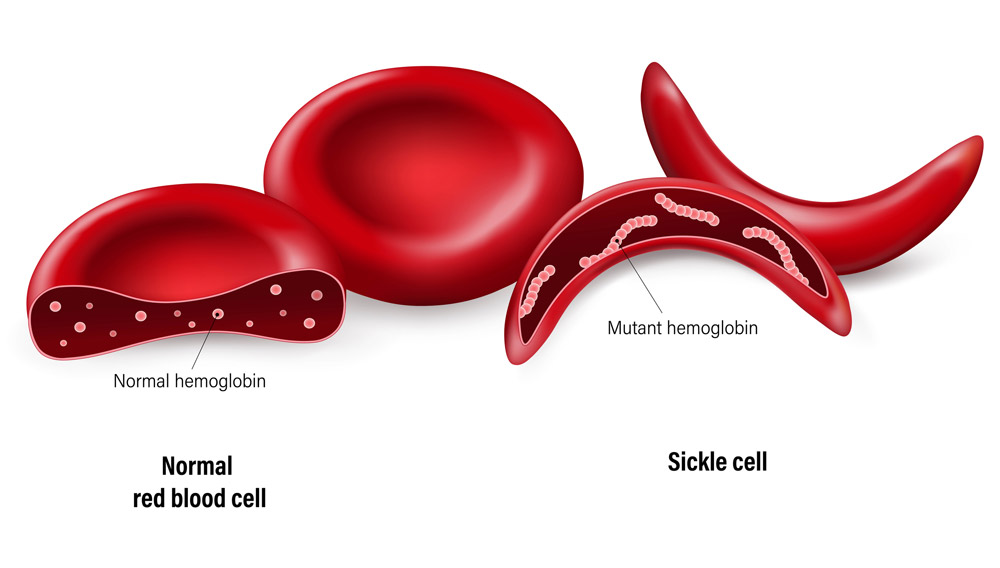
Schon seit einigen Jahren erproben Wissenschaftler auch den Einsatz der Genschere CRISPR/Cas9 gegen beide Blutkrankheiten. Bereits 2016 gelang es einem Team, den Gendefekt der Sichelzellanämie in menschlichen Blutstammzellen zu reparieren – allerdings nur im Labor und mit anschließender Re-Implantation in Mäuse. 2015 versuchten chinesische Forscher, den Thalassämie-Defekt bei menschlichen Embryos zu reparieren – mit eher geringem Erfolg. Weil dies jedoch einen ethisch umstrittenen Eingriff in die menschliche Keimbahn darstellt, sorgte dies weltweit für Kritik.
Gentherapie reaktiviert fötales Hämoglobin
Jetzt hat die britische Arzneimittelbehörde MHRA erstmals eine Gentherapie beider Blutkrankheiten zugelassen, die auf CRISPR/Cas9 beruht. Die von den Unternehmen Vertex Pharmaceuticals und CRISPR Therapeutics entwickelte Behandlung ist weltweit die erste Zulassung einer Gentherapie mit der Genschere. „Ich bin erfreut, verkünden zu können, dass wir eine innovative und bisher einmalige Gentherapie namens Casgevy zugelassen haben“, sagt Julian Beach von der MHRA.
Allerdings repariert die Casgevy-Gentherapie nicht den Gendefekt, der die Blutkrankheiten verursacht. Stattdessen reaktiviert sie die Bildung eines fötalen Hämoglobins, das normalerweise nur bei Kindern im Mutterleib und bei Neugeborenen produziert wird. Die CRISPR/Cas-Therapie erreicht dies, indem ein normalerweise für den Stopp der fötalen Produktion zuständiges Gen außer Funktion gesetzt wird. Dadurch kann das fötale Hämoglobin die Funktion des krankhaften Blutfarbstoffs übernehmen und die Blutarmut kurieren.
Aufwendige Prozedur
Die Zulassung der Casgevy-Gentherapie beruht auf klinischen Studien, bei denen 45 Patienten mit Sichelzellanämie und 54 Patienten mit Beta-Thalassämie behandelt wurden. Dafür wurden ihnen zunächst Blutstammzellen aus dem Knochenmark entnommen, bei denen dann im Labor das entsprechende Gen mit CRISPR/Cas9 verändert wurde. Währenddessen müssen sich die Patienten einer Chemotherapie unterziehen, die die krankhaften Blutstammzellen in ihrem Knochenmark weitgehend abtötet, damit die geneditierten Stammzellen Platz zum Ansiedeln haben.
Anschließend erhalten die Patienten ihre geneditierten Blutstammzellen mittels Infusion wieder zurück. Diese vermehren sich dann im Knochenmark und bilden rote Blutkörperchen, die einen hohen Anteil fötales Hämoglobin enthalten. In den Zulassungsstudien waren dadurch 28 der 29 Sichelzell-Patienten, die die Studie beendeten, schmerzfrei und benötigten keine Bluttransfusion. Bei der Thalassämie konnten 39 von 42 Patienten ganz auf Bluttransfusionen verzichten, die restlichen benötigten nur noch ein Drittel der früheren Menge.
Zulassungsbehörde und Unternehmen gehen davon aus, dass die Gentherapie langfristig wirkt. Wie lange die Wirkung jedoch anhält und ob sie vielleicht doch wiederholt werden muss, ist bisher unbekannt.
Kein Ersatz für bisherige Therapien, aber eine Ergänzung
„Der große Vorteil der CRISPR-Therapie ist, dass es anders als bei der Stammzelltransplantation weder eine Immunreaktion des Körpers, die zu einer Abstoßung führt, noch eine Immunreaktion des Spenders gibt. Denn es sind ja die eigenen Zellen, die zurück in den Körper kommen“, erklärt der nicht an den Studien beteiligte Onkologe Selim Corbacioglu vom Universitätsklinikum Regensburg. Allerdings wird der eigentliche Gendefekt der beiden Erbkrankheiten nicht repariert, sodass die behandelten Patienten diesen noch immer an ihren Nachkommen weitergeben können.
Hinzu kommt: Die Gentherapie mittels Casgevy ist für die Patienten ähnlich belastend wie eine Knochenmarksspende und Stammzelltherapie. „Zudem ist die Therapie sehr teuer: Mehr als zwei Millionen Euro soll sie pro Patient kosten. Die Stammzelltransplantation liegt bei maximal 300.000 Euro“, sagt Corbacioglu. Das sei für die Gesundheitssysteme nur schwer zu stemmen. „Die CRISPR-Therapie wird daher auf absehbare Zeit nicht die Stammzelltransplantation ersetzen, sondern sie ergänzen.“
Auch in anderen Ländern läuft das Zulassungsverfahren für die CRISPR-Therapie: In den USA soll die Entscheidung über die Zulassung von Casgevy gegen Sichelzellanämie Anfang Dezember fällen, für Beta-Thalassämie im Frühjahr 2024. Die europäische Arzneimittelbehörde EMA hat ebenfalls bereits mit der Prüfung der Casgevy-Gentherapie begonnen.
Quelle: Medicines and Healthcare products Regulatory Agency (MHRA), Science Media Center