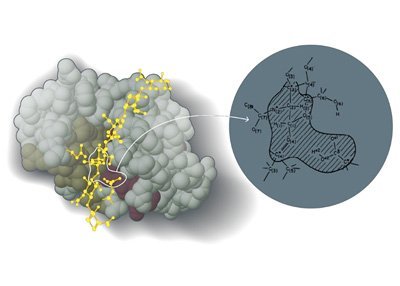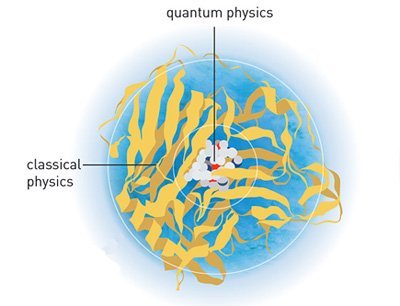
Den Chemie-Nobelpreis erhalten drei Forscher, die die Grundlage legten für etwas, das uns heute selbstverständlich erscheint: Dreidimensionale Computersimulationen von chemischen Molekülen und ihren Reaktionen. Martin Karplus, Michael Levitt und Arieh Warshel entwickelten in den 1970er Jahren erstmals einen Weg, um die komplexen Vorgänge an der Grenze zwischen klassischer Chemie und Quantenphysik per Computer zu modellieren.
Moderne Chemie findet heute längst nicht mehr nur im Reagenzglas statt. Vieles geschieht inzwischen am Rechner. Denn chemische Reaktionen laufen innerhalb von Sekundenbruchteilen ab: Blitzschnell lagern sich Atome um, bilden sich Bindungen oder zerfallen ganze Molekülketten in ihre Einzelteile. Das Ganze passiert viel zu schnell, um es direkt beobachten zu können. Wenn es beispielsweise darum geht, den komplexen Ablauf bei der Photosynthese einer Pflanzen zu entschlüsseln oder die Funktionsweise eines Enzyms, kommen daher auch Chemiker nicht mehr ohne Modelle der Moleküle und Reaktionen aus.
Schwer fassbare Elektronen
Doch lange Zeit war das nur statisch möglich: Die Programme schafften es, die beteiligten Moleküle in ihrem Grundzustand dreidimensional abzubilden. Dadurch bekamen die Forscher zumindest einen Eindruck davon, wie die Atome angeordnet waren. Das Problem dabei: Während einer chemischen Reaktion verändert sich nicht nur der Bindungszustand des Gesamtmoleküls, auch die Atome verlassen ihren Grundzustand und nehmen beispielsweise Energie auf. Dadurch verändern sich die Bahnen der Elektronen in ihren Orbitalen – den entscheidenden Akteur für die chemischen Bindungen.
Doch genau hier beginnt das Problem: Elektronen sind schwer fassbar – und damit auch schwer abzubilden oder zu modellieren. Als Partikel, die sowohl Eigenschaften von Teilchen als auch von Wellen haben, gehorchen sie den Gesetzen der Quantenphysik. Wo sich ein Elektron gerade befindet, lässt sich daher nur in Wahrscheinlichkeiten ausdrücken. Das aber macht ihre Simulation extrem aufwändig und kostet enorme Rechenleistung. In den 1970er Jahren war es daher nur möglich, die Konfiguration sehr kleiner und simpler Moleküle zu simulieren. An diesem Punkt kommen nun die drei Nobelpreisträger ins Spiel.